Post-Mendelian Genetic Model in COVID-19
Nicola Picchiotti1,2#, Elisa Benetti3#, Chiara Fallerini3,4#, Sergio Daga3,4, Margherita Baldassarri3,4, Francesca Fava3,4,5, Kristina Zguro3, Floriana Valentino3,4, Gabriella Doddato3,4, Annarita Giliberti3,4, Rossella Tita5, Sara Amitrano5, Mirella Bruttini3,4,5, Laura Di Sarno3,4, Diana Alaverdian3,4, Giada Beligni3,4, Maria Palmieri3,4, Susanna Croci3,4, Mirjam Lista3,4, Ilaria Meloni3,4, Anna Maria Pinto5, Chiara Gabbi6, Stefano Ceri7, Antonio Esposito7, Pietro Pinoli7, Francis P. Crawley8, Elisa Frullanti 3,4, Francesca Mari3,4,5, GEN-COVID Multicenter Study, Marco Gori1,9, Alessandra Renieri3,4,5*, Simone Furini3*
1University of Siena, DIISM-SAILAB, Siena, Italy
2Department of Mathematics, University of Pavia, Pavia, Italy
3Med Biotech Hub and Competence Center, Department of Medical Biotechnologies, University of Siena, Italy
4Medical Genetics, University of Siena, Italy
5Genetica Medica, Azienda Ospedaliero-Universitaria Senese, Italy
6Department of Biosciences and Nutrition, Karolinska Institutet, Stockholm, Sweden
7Politecnico di Milano, DEIB, Milano, Italy
8Good Clinical Practice Alliance-Europe (GCPA) and Strategic Initiative for Developing Capacity in Ethical Review-Europe (SIDCER), Leuven, Belgium.
9Universite Côte d’Azur, Inria, CNRS, I3S, Maasai
*Corresponding author: Alessandra Renieri, Medical Genetics Unit, University of Siena, Viale Bracci, 253100 Siena, Italy
Simone Furini, Department of Medical Biotechnologies, University of Siena, Le Scotte Polyclinic-Administrative Building,Viale Bracci, 253100 Siena, Italy
Received: 02 November 2021; Accepted: 10 November 2021; Published: 09 December 2021
Article Information
Citation:
Picchiotti N, Benetti E, Fallerini C, Daga S, Baldassarri M, Fava F, Zguro K, Valentino F, Doddato G, Giliberti A, Tita R, Amitrano S, Bruttini M, Sarno LD, Alaverdian D, Beligni G, Palmieri M, Croci S, Lista M, Meloni I, Pinto AM, Gabbi C, Ceri S, Esposito A, Pinoli P, Crawley FP, Frullanti E, Mari F, GEN-COVID Multicenter Study, Gori M, Renieri A, Furini S. Post-Mendelian Genetic Model in COVID-19. Cardiology and Cardiovascular Medicine 5 (2021): 673-694.
View / Download Pdf Share at FacebookAbstract
Host genetics is an emerging theme in COVID-19. A handful of common polymorphisms and some rare variants have been identified, either through GWAS or candidate gene approach, respectively. However, an organic model is still missing. Here, we propose a model that takes into account both common and rare coding variants. This model has been piloted in a cohort of 1,318 Italian SARS-CoV-2 positive individuals. Ordered logistic regression of clinical WHO grading on age, stratified by sex, was used to obtain a binary phenotypic classification useful for subsequent data mining. Genetic variability from WES was synthesized in several Boolean representations differentiated according to allele frequencies and genotype effect. LASSO logistic regression was used for extracting relevant features, part of them being sex specific. We defined a set of common coding polymorphisms relevant for COVID-19 severity. Another set of rare coding variants were found to contribute either to mildness or severity of COVID-19, which in some cases simulate a Mendelian inheritance. The combined effect of common and rare variants can be described as an Integrated PolyGenic Score (IPGS). This score can be computed using the following formula: (nmildness-nseverity) +F (mmildness-mseverity). Here n is the number of common driver genes, m is the number of driver rare variants, and F is a factor for an appropriate weighing of the more powerful rare variants. We named this model: “post-Mendelian”. The model has been tested with multiple train-test splits and the polygenic score accurately separated patients with severe COVID-19 from those having a mild form. Furthermore, segregation of IPGS can be observed in familial cases in which more than one infected individual is present.
Keywords
<p>COVID-19; Genetic science modelling; Host genetics; Integrative polygenic score</p>
Article Details
1. Introduction
Coronavirus disease 2019 (COVID-19) presents an important test case for developing new models for studying complex disorders with a background of combined genetic and environmental factors. Unlike other multifactorial disorders, the main environmental factor for COVID-19, SARS-CoV-2, can easily be identified through PCR-based tests on swab. Assuming a relatively low impact of viral genome variability [1], the remaining variability in clinical outcome may likely be associated with age and host genetics, including sex. Genome-Wide Association Studies (GWASs) have identified a certain number of common polymorphisms in relevant genes [2-4]. However, these associations do not fully explain the variability of clinical outcomes. The candidate gene approach has shown that, as with many other complex disorders, a simple Mendelian inheritance is also found in COVID-19, affecting some rare individuals with defective gene variants related to innate immunity [5-7].
In early COVID-19 research, the Italian GEN-COVID Multicenter Study explored COVID-19 host genetics through Whole Exome Sequencing analysis (WES) in a cohort of 35 hospitalized patients. This research led to the preliminary identification of a combination of rare and common variants that appeared to potentially impact patient clinical outcome [8]. Then, within this Italian GEN-COVID Multicenter Study, biospecimens from more than 1,000 SARS-CoV-2 positive individuals were collected in the GEN-COVID Biobank (GCB), with clinical data stored in the related Patient Registry (GCPR), and genetic data in the connected Genetic Data Repository (GCGDR) [9]. SNP genotyping of this cohort contributed to the identification of some loci associated with COVID-19 [3], while WES analysis pinpointed additional common non bi-allelic polymorphisms [10-12] and rare coding variants [13]. However, an organic model explaining how common variants may combine with rare variants is still lacking. In the study presented in this paper, we propose a new model for describing the severity of COVID-19 using both common and rare coding variants. The model was defined in two steps: in the first step, logistic regression approach was used to identify a set of genes that are predictive for the severe or the mild phenotype of COVID-19. In the second step of the model, these extracted predictive genes were used to define a score that separates the extreme COVID-19 phenotypes, giving a disease severity prediction.
2. Materials and Methods
2.1 Patient’s cohort and clinical classification
Demographic and clinical characteristics of the data set were identified and published [9]. The number of cases used for this study was 1,318. In order to obtain a clinical classification as independent as possible from age and sex, an Ordered Logistic Regression (OLR) model was used. Separately for the male and female cohorts, two OLR models were fitted using age to predict the ordinal grading (0, 1, 2, 3, 4, 5) dependent variable (Figure 1). Then, each patient was assigned a clinical classification: equal to 0 (mild), if the actual patient grading was below the one predicted by the OLR; or equal to 1 (severe), if the grading was above the OLR prediction. The patients with a predicted grading equal to the actual grading were excluded from the following analyses in order to compare patients where the genetic contribution towards the severe/mild phenotype is expected to be more relevant.

Figure 1: Clinical classification adjusted by age. Two Ordered Logistic Regression (OLR) models, stratified by sex, fitted using age to predict the ordinal grading (0, 1, 2, 3, 4, 5) dependent variable. On the Y axis, the grading according to patients’ treatment is reported (5=deceased; 4=intubated; 3=CPAP/biPAP; 2=oxygen therapy; 1=hospitalized without oxygen support; 0=not hospitalized oligo-asymptomatic patients). On the X axis, age is reported. Red dots represent subjects falling above the expected treatment outcomes according to age (hence considered severe), green dots are subjects falling below the expected treatment outcomes according to age (hence considered mild) and black dots are subjects matching the expected treatment outcomes according to age (hence considered intermediate).
2.2 WES analysis
Whole Exome Sequencing with at least 97% coverage at 20x was performed using the Illumina NovaSeq6000 System (Illumina, San Diego, CA, USA). Library preparation was performed using the Illumina Exome Panel (Illumina) according to the manufacturer's protocol. Library enrichment was tested by qPCR, and the size distribution and concentration were determined using Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). The Novaseq6000 System (Illumina) was used for DNA sequencing through 150 bp paired-end reads. Variant calling was performed according to the GATK4 best practice guidelines, using BWA for mapping and ANNOVAR for annotating. WES data were represented in a binary mode on a gene-by-gene basis.
2.3 Boolean representation of bi-allelic polymorphisms
In the Boolean representation of bi-allelic polymorphisms, coding variants (missense, non-sense, and ins/del), as well as splicing mutations, with a frequency > 1% as defined in gnomAD for the Non-Finnish European population were considered. Depending on the boolean representation considered (see section 2.6) only homozygous variants, or both homozygous and heterozygous variants were considered. For those genes having more than one common variant, all the unique combinations (mutually exclusive) with a frequency above 5% in the cohort were represented. For instance, IBSP gene has 5 coding polymorphisms with 5 combinations and with a frequency above 5% in the cohort. Each combination was represented separately as IBSP_1 to IBSP_5. A further feature, named IBSP_0, was defined to represent the presence of any combination with a frequency lower than 5%. These unique combinations were used to define a matrix of M by N input features (M and N being, respectively, the number of combinations and the number of samples), with the element j,i equal to 1 if the combination of common variants j is present in the sample i.
2.4 Boolean representation of rare variants
Three models were proposed for the binary representation of rare variants: autosomal dominant (AD), autosomal recessive (AR), and X-linked (XL). Only coding variants (missense, non-sense, and ins/del), as well as splicing mutations, were considered, together with any pathogenic mutations (coding and non-coding) having a frequency below 5% (e.g. Phe508del in CFTR being 1.1% in the non-Finnish European population). In the AD model, the input feature j,i was 1 if gene j in sample i had at least one variant with a frequency ≤ 1%, as defined in gnomAD for the Non-Finnish European population. In the AR model, the input feature j,i was 1 if gene j in sample i had either a variant with a frequency ≤ 1% in a homozygous state or two variants. In the XL model, only genes on chromosomes X were considered. The input feature j,i is 1 if gene j in sample i has a variant with frequency ≤ 1% (hemizygous state in males).
2.5 Sample pre-processing
Among the 1,318 samples, after PCA analysis, 63 outliers were removed. Those corresponded to non-white subjects (32 hispanic, 12 black, 18 asian, 1 arabic). In the Boolean representation of rare variants, the cumulative probability distribution for the number of mutated individuals per gene was estimated, and the elbow method was used to identify the number of mutated individuals per gene that corresponds to a change in the convexity of the cumulative probability. The genes with a number of mutated individuals above this threshold were considered artefacts and not included in further analyses. Among the 18,085 annotated genes, 18 genes were excluded from the analysis due to annotation errors, such as the ARSD and the VCX family genes.
2.6 LASSO logistic regression
The binary classification problem, i.e., mild/severe cases, was solved by a logistic regression model, one of the most common and successful Machine Learning (ML) algorithms for binary classification tasks with probabilistic interpretation. For each sex, 6 separate logistic models were defined using the following input features: 1) common bi-allelic coding haplotypes of autosomal genes (hetero plus homo versus wt) extracting common variants acting in a heterozygous state; 2) common bi-allelic coding haplotypes of autosomal genes (homo versus hetero and wt) extracting common variants acting in a homozygous state; 3) common bi-allelic coding haplotypes of X-linked genes extracting common variants acting in a hemizygous state in males or as X-linked dominat in females; 4) rare variants of autosomal genes (hetero plus homo versus wt) extracting rare variants acting in heterozygous state; 5) rare variants of autosomal genes (homo versus hetero and wt) extracting rare variants acting in a homozygous state; and 6) rare variants of X-linked genes extracting rare variants acting in a hemizygous state in males or as X-linked dominat in females. In order to enforce both the sparsity and the interpretability of the results, the model was trained with the additional LASSO (Least Absolute Shrinkage and Selection Operator) regularization term. By denoting with the coefficients of the logistic regression model, and by denoting with lambda (λ) the strength of the regularization, the LASSO regularization term of the loss 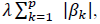 has the effect of shrinking the estimated coefficients to 0, providing a feature selection method for sparse solutions within the classification task. The weights resulting from the logistic regression algorithm can be interpreted as the feature importances for the task [14].
has the effect of shrinking the estimated coefficients to 0, providing a feature selection method for sparse solutions within the classification task. The weights resulting from the logistic regression algorithm can be interpreted as the feature importances for the task [14].
The fundamental hyper-parameter of the logistic regression algorithm is the strength of the LASSO term, which was tuned with a grid search method on the average accuracy for the 10-fold cross-validation. 50 equally spaced values in logarithmic scale in the range 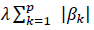 were tested. The optimal regularization parameter was selected as the one with the best trade-off between the simplicity of the model and the cross-validation score, i.e., as the highest value providing an average score close to the highest score with its half standard deviation. The method is designed to select the most important genes (and not necessarily the entire set of genes contributing to COVID-19 variability). Data preprocessing was coded in Python. For the logistic regression model, the scikit-learn module with the liblinear coordinate descent optimization algorithm was used. The entire cohort was splitted into a training set (90%) and a testing set (10%). The model has been tested with multiple train-test splits.
were tested. The optimal regularization parameter was selected as the one with the best trade-off between the simplicity of the model and the cross-validation score, i.e., as the highest value providing an average score close to the highest score with its half standard deviation. The method is designed to select the most important genes (and not necessarily the entire set of genes contributing to COVID-19 variability). Data preprocessing was coded in Python. For the logistic regression model, the scikit-learn module with the liblinear coordinate descent optimization algorithm was used. The entire cohort was splitted into a training set (90%) and a testing set (10%). The model has been tested with multiple train-test splits.
2.7 The post-Mendelian model
The proposed model aims at combining the genetic knowledge extracted from the individual LASSO logistic regression models by extending the standard “threshold model” of common polymorphisms to a more general framework, including the effect(s) of rare variants. The main hypothesis of the model is that it is possible to define an Integrated PolyGenic core (IPGS) by counting the number of variants in genes promoting the severe or the mild phenotype, assigning a different weight to common and rare variants.
The following formula calculates the IPGS as:
IPGS = (n_mildness - n_severity) + F (m_mildness - m_severity)
In the equation above, n_mildness and n_severity are the number of common polymorphic driver genes conferring mildness or severity (respectively) to COVID-19; while m_mildness and m_severity are the number of driver rare variants conferring severity or mildness (respectively) to COVID-19 (Figure 2).

Figure 2: The determinative formula for the post-Mendelian model. This formula determines the integrated polygenic score (IPGS). The first term is the difference between the number of common variants/coding haplotypes conferring mildness (n_mildness) and severity (n_severity) to COVID-19; whereas the second term is the difference between the number of rare variants conferring mildness (m_severity) and severity (m_severity) to COVID-19. The multiplicative factor F was included to model the penetrant effect of rare variants in comparison to common variants.
The genes conferring mildness or severity were identified using the LASSO logistic regression models described in section 2.6. Boolean representations 1-3 were used to define n_mildness and n_severity, and boolean representations 4-6 were used to define m_mildness and m_severity. IPGS value calculated for each sex excludes genes specific of the other sex. In order to identify the optimal value of the F factor, for each value of F in the range 0-5, the silhouette coefficient of the clustering (mild vs severe) results was calculated. The optimal value of F was defined as the one that maximizes the silhouette coefficient, i.e., provides the best separation between the two clusters. The entire procedure is schematized in Figure 3.

Figure 3: Outline of the method. Separately for the male and female cohorts, two OLR models were fitted using the age to predict the ordinal grading (0, 1, 2, 3, 4, 5) dependent variable. Then, each patient was provided a clinical classification equal to 0 (green), if the actual patient grading was below the one predicted by the OLR, or 1 (red), if the actual patient grading was above the OLR prediction. The patients with a predicted grading equal to the actual grading (black) were excluded from the LASSO analysis. LASSO Logistic Regression was performed on WES data represented in a Boolean manner, including common and rare variants, separately for males and females. Following the analysis provided on the basis of the WES data analyzed through LASSO Logistic Regression, the post-Mendelian model was applied. For each subject then, an integrated polygenic score (IPGS) was calculated.
2.8 Bootstrap analyses
In order to have more robust feature selection the dataset was divided into a training set and a testing set (90/10), and the feature selection was performed using only samples in the training set. A bootstrap approach with 82 iterations was adopted (Supplementary Tables 1-4). At each bootstrap iteration, 90% of the samples were selected (without replication), to identify the most relevant features for each Boolean representations (Supplementary Tables 1-4).
3. Results
3.1 Assessing clinical classification stratified by sex and adjusted by age
Whole Exome Sequencing (WES) data stored in the Genetic Data Repository of the GEN-COVID Multicenter Study (GCGDR) and coming from biospecimens of 1,318 SARS-CoV-2 PCR positive subjects were used for the analysis [9]. Because age and sex are strong determinants of the clinical outcome, we stratified the cohort by sex and then applied ordered logistic regression to re-classify the patients (Figure 1) as follows: i) severe: subjects falling above the expected treatment outcomes according to age; ii) intermediate: subjects matching the expected treatment outcomes according to age; iii) mild: subjects falling below the expected treatment outcomes according to age. This novel classification is expected to be reliable independent of age. If this is further demonstrated, it should then facilitate the reliable identification of genetic factors responsible for COVID-19 severity as well as potentially identifying (druggable) pathways to treatment.
3.2 The advantage of using several Boolean representations
We reasoned that, in order to identify possible Mendelian-like COVID-19 disorders, different rare variants in the same gene be classified as 1 or 0 (0 being the absence of variants). In order to detect the underlying Mendelian-like mode of inheritance, genes on chr X were considered separately. This representation is able to extract X-linked recessive genes on males (Figure S1q), such as TLR7 which is now functionally validated by three independent studies [6,7,13].
Likewise, in order to distinguish autosomal dominant from autosomal recessive disorders, two different Boolean representations were considered for the remaining autosomal genes: at least one variant equal for the dominant model and at least 2 variants for the recessive models (Figures S1i-S1p). Furthermore, we reasoned that for the contribution of common polymorphisms, based on the chromosomal location (X versus autosomal) and the genotype (at least one variant versus at least 2 variants), three different Boolean representations were better suited to identify common variants (Figures S1a-S1h). These representations were able to extract autosomal genes acting on specific sex with a specific genotype. For example, using at least one variant in males, LASSO pinpoints the p.Leu412Phe polymorphism in the testosteron-repressed TLR3 gene (Figure S1b). This is an already known functional polymorphism [15], which is now associated to COVID-19 by two independent studies [11, 16], proved to reduce TNF alpha and autophagy [11].
3.3 Discovery of genes and sex dependent effect
We used severe versus mild cases defined by ordered logistic regression of the training set as inputs for a series of logistic regression models using LASSO regularization. The purpose of these logistic models was to identify which features are better predictors of either severe or mild disease. The six different types of genetic variability identified above were represented in a Boolean manner and tested separately. The full list of genes identified in both sexes are reported in Supplementary Tables 1-4.
Using the cohort inclusive of both sexes, for common bi-allelic haplotypes (hetero plus homo versus wt), we identified a set of genes ordered by importance (Figure 4). We then repeated the analysis using sex stratification (Figure 4). We found that a part of them are sex-specific, i.e., found in one sex only (Figure S1a, Figure S1b and S1c, Supplementary Tables 1-4). A similar pattern was found for all other Boolean representations.

Figure 4: Sex specific effect. Upper panel: genes with common bi-allelic haplotypes (hetero plus homo versus wt), ordered by importance in the cohort that includes both sexes. Down panel: genes with common bi-allelic haplotypes (hetero plus homo versus wt), ordered by importance and stratified by sex (left side: genes in males; right side: genes in females).
3.4 Discovery of relevant genes with common variants
Applying LASSO logistic regression to the Boolean representation of common bi-allelic haplotypes, we identified a set of relevant genes for COVID-19 severity (Figure S1a-S1h and Supplementary Tables 1 and 2). Many genes were found in both sexes (Figure S1a, S1d and Supplementary Tables 1 and 2) and a set of them only affect one sex (Figure S1b, S1c, S1e, S1f, S1g, S1h and Supplementary Tables 1 and 2). Among the relevant common variants associated with severe disease, there are some genes already functionally validated and reported associate with severe COVID-19 disease such as TLR3 with p.Leu412Phe polymorphism in heterozygous state [11,16] that controls interferon response; and SELP with p.Asp603Asn polymorphism in homozygous state [12], a cell adhesion molecule which mediates the activation of platelets and endothelial cells and its soluble isoform is increased in both venous and arterial thrombosis. Both TLR3 and SELP are testosterone regulated genes and in fact impact only on males patients.
3.5 Discovery of relevant genes with rare variants
Using LASSO logistic regression on the Boolean representation of rare variants, we identified a set of relevant genes for COVID-19 severity (Figure S1i-S1r, Supplementary Tables 3 and 4). Many genes were found in common with both sexes (Figure S1i, S1n and Supplementary Tables 3 and 4). Among the relevant genes extracted only in males with rare variants associated with severe disease, there is the already known TLR7 involved in controlling interferon response [6,7,13]. Among relevant genes with rare variants associated with severe diseases in females there is TLR5 (hetero) previously involved in bacterial infection and more recently also in viral infection.
3.6 The post-Mendelian paradigm for COVID-19 modelization
In the previous sections, the protocol used to identify predictive genes for the severe and mild phenotypes were presented and the plausibility of these genes is supported by biological evidence. This agreement leads to plausible biological knowledge based on two independent modelling approaches. The apparent strong confirmation of our hypothesis convinced us to further explore the possibility of combining the derived variants' information regarding these genes into a predictive model of phenotype expression. The proposed model extends the standard “threshold model” of common polymorphisms to a more general framework, including both the effect of common and rare variants. In the case that each common variant carries the same relative risk, subjects with approximately the same number of common variants in genes indicative of mild or severe will behave according to their age (recall the black dots in the ordered logistic regression of Figure 1). In other cases, subjects with unbalanced variants (that is, with a higher number of mild- or severe disease gene variants) will more likely belong to the corresponding phenotype, with a probability that increases as the difference between the number of mild- and severe gene variants increases. By looking at the epidemiological data of the total number of those who are severely ill (intubated or CPAP-BiPAP), one should be able to infer the exact number of genes involved in the model and the exact percentage necessary for the threshold effect. In addition to the cumulative effect of common variants, another group of genes may be affected by a rare mutation. According to an autosomal dominant, autosomal recessive, or X linked mode let us suppose a rare mutation occurring in either a neutral or severe background of common variants. In such cases, a more severe or earlier disease segregating as a Mendelian inheritance may be identifiable. Family members are also likely to have identical or similar combinations of common variants. If a rare variant is identified against a mild disease background, the penetrability of the virus may likely be dependent on the strength of the rare variant in warding off viral replication. If a mildness rare occurs in a neutral or mildness background, the individual will be even more likely to exhibit a mild phenotype (for example, an individual with the persistence of antibodies far away from the vaccine or infection). If rare mildness variants occur in a harmful background, its effect will likely depend on the strength (relevance) of mildness (the type of gene and its mutation as well as the location of the gene in the pathophysiological process. For example, individuals with rare variants in FACL4 (which prevents the accumulation of lipid microparticles necessary for the development of the virus) could have milder symptoms even in a negative background (because the action is upstream). On the other hand, some individuals are asymptomatic following infection but continue to test positive for SARS-CoV-2 for a significant period without developing COVID-19. These could be individuals with common mildness variants but “severity rare mutations” favoring the spread of the virus on the tissues. However, the downstream process appears milder because they respond well by innate and adaptive immunity. The number of rare variants that individuals in the pilot study were found to have varied from 0 to 10, with an average of 3. Some individuals without rare variants were also found to present with or develop severe disease, indicating that common variants alone could be involved in affecting disease severity. In individuals with rare variants, the correct prediction of disease severity required a consideration of both common variants and rare variants. In cases of individuals having more than one rare variant gene in the same variant category (e.g., mild or severe), the segregation gene variants simulated the oligogenic model (digenic, trigenic, etc.).
3.7 Fitting the model in the cohort and calibration of rare variants contribution
In the cohort of 1,318 SARS-CoV-2 used in the pilot study, we used the post-Mendelian model depicted in the previous section to determine an integrated polygenic score in which rare variants contribute together with common polymorphisms to COVID-19 liability. By counting the mutations of genes extracted by LASSO logistic regression for each patient in the cohort, we were able to obtain the integrated polygenic scores as reported in the frequency distribution shown in Figure 5 separately for severe (red) and mild (green) phenotypes. The calibrated F factor (i.e. the coefficient for which the mean silhouette coefficient is at its highest) is shown in Figure 5 for the male (2) and female (1.2) cohorts. In the overall cohort (male and female cohorts taken together), classical Mendelian inheritance was simulated when rare variants occurred in a medium or severe background (subjects having only 1 susceptibility mutation) and oligogenic inheritance (having 2 susceptibility mutations, having 3 mutations, etc.).

Figure 5: Integrated Polygenic Score, factor calibration, and phenotypes. Frequency distributions of the IPGS score for the male cohort (Panel a) and female cohort (Panel b). The red distribution is related to the severe phenotypes and the green one to the mild phenotypes. Panel c and Panel d report the mean silhouette coefficient of the clustering of mild and severe phenotypes as a function of possible F values of the IPGS formula in the range 0-5. The coefficient is a measure of the goodness of the clustering. The maximum value that provides the best separation between the two clusters is 2 for the male cohort (Panel c) and 1.2 for the female cohort (Panel d).
3.8 Segregation of the post-Mendelian model in families
In the overall cohort 15 familial cases were collected and analysed. Segregation analysis using an integrated polygenic score is in line with the phenotype in each of the families (Figure 6). Through further exploration for rare variants in this individual family members, an extremely rare pathogenic mutation was detected in IFNAR1. The frequency, however, of IFNAR1 variants was too low to identify using logistic regression in a cohort of this size.

Figure 6: Segregation analysis afforded by the post-Mendelian model. Example of segregation analysis using the post-Mendelian model applying an integrated polygenic score to 9 pedigrees. The squares represent male subjects and the circles represent female subjects. Red = severely affected (category 1 in Figure 1); green = oligo-asymptomatic subjects (category 0 in fig. 1); grey squares represent intermediate subjects (category black in Figure 1). Under each symbol is reported the treatment and (in parenthesis) the age. For each patient the formula for the computation of the integrated polygenic score (IPGS) is reported. Panel a: Brothers of 32 and 31 years with discordant phenotypes: hospitalized CPAP treated, and oligosymptomatic, respectively. In agreement with their phenotype, they have IPGS 0 and 10.1 respectively, mainly due to increased common polymorphisms associated with mild disease in the asymptomatic brother (such as p.Ile57Val of AURKA, a cell cycle regulator downregulated during SARS-CoV-2 infection; p.Cys357Arg/p.Va335Met of GBP3, a strong repressor of the activity of the viral polymerase complex, which results in decreased synthesis of viral proteins; and p.M1? of TLR8, a member of the Toll-like receptor family which plays a fundamental role in pathogen recognition and activation of innate immunity). Panel b: Sisters of 62 and 60 years of age with partially discordant phenotype, hospitalized with oxygen support only and hospitalized CPAP treated, respectively. In agreement with their phenotype, they have IPGS minus 2.8 and minus 7.05, respectively, mainly due to increased severity of rare variants in the more severely affected sister (including Amyloid Beta Precursor Protein Binding Family A Member 3 APBA3, which plays a role in immune response; and the low density lipoprotein receptor family member LRP8, which has a role in the suppression of innate response). Treatment with immunosuppressive agents may be an option in this patient.
3.9 Testing the model
In order to test whether the proposed model is able to generalize the predictions to unseen samples, the entire cohort was splitted into a training set and a testing set (10%). The entire training procedure was completely blind to the samples in the testing set, namely, the training set was used to: (i) train the OLR models for predicting the phenotype adjusted by age and sex; (ii) identify the parameters and the regularization strength (by cross-validation on the training set) of the LASSO logistic regression models; (iii) estimate the optimal value of the F score in the IPGS. The predictivity of the model was estimated by using the ROC-AUC. In order to have a robust estimate of the predictivity of the model, the whole procedure of train-test split was repeated 150 times. Then, the distribution of the ROC-AUC scores was compared with the one (null-hypothesis) obtained by the shuffling of the phenotypes. As expected, the average ROC-AUC is centered at 0.5 in the presence of random phenotype. Instead, when the actual phenotypes are adopted the ROC-AUC distribution is significantly shifted towards higher values (p-value of 2.7x10-6) as shown in Figure 7.

Figure 7: Model performances. The ROC-AUC is compared between models trained using randomly shuffled phenotypes (blue boxes) and the actual COVID-19 severity phenotypes (orange boxes). The two-sided t-test for the null hypothesis that 2 independent samples have equal average is performed with a statistically significant p-value of 2.7x10-6.
4. Discussion
Using data analysis techniques to investigate approximately 20,000 genes, we have identified a subset of a few hundred candidate genes involved in the COVID-19 mild and severe disease. Some of those candidate genes appear to be involved in disease severity when associated with rare variants; while others of them appear to be involved though one or more common polymorphism/s.
Extracted genes bearing rare variants simulate a Mendelian-like disorder in infected members. Among these is the recently identified X-linked recessive disorder associated with the TLR7 gene [6, 7, 13]. In this case males appear to be severely affected due to a defect in the viral sensor TLR7, which is unable to appropriately trigger the downstream events leading to interferon production. The appropriate therapy for these males is interferon as soon as the infection is identified. About ten families with this defect in the cohort of the pilot study were identified. Among females an autosomal dominant-like disorder conditioned by sex is due to TLR5, another sensor, previously involved in bacterial infection and more recently also in viral infection [17, 18]. This is an autosomal gene inhibited by estrogen. Those who inherit the mutation are at significant risk of severe COVID-19, especially if they are females in which the amount of the protein is naturally reduced. The recommended treatment, as with TLR7, is interferon. Some tens of families with this variant were present in the cohort of the pilot study. There are also inverted Mendelian disorders leading to inheritance of protective rare variants. These include: i) the XPNPEP2 gene, the ACE2 co-receptor that is also involved in the metabolism of bradykinin, a potent vasodilator that acts as a protective against vasoconstriction and thrombosis [19]. The gene is found on the X chromosome, thus having a role in protecting males; and ii) VEGFD gene, encoding for vascular endothelial growth factor D, predominantly expressed in the lungs, interacting with ACE2, with an, important role in the pathogenesis of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) by its properties to increase vascular permeability [20,21]. Extracted genes bearing common variants delineate different groups of “pathogenicity”. For example, TLR3 p.Leu412Phe (MAF 29,79%) confers reduced inflammatory response (reduced TNFa) and reduced autophagy [11]. The effect is enhanced in males because the TLR3 is down regulated by testosterone. In these patients hydroxychloroquine, which inhibits autophagy, is particularly dangerous. Most importantly, we defined a new genetic method for modelling COVID-19 severity (Figure 2). The proposed method has a number of innovative approaches. To arrive at this model based on this formula, first the clinically modified WHO outcome scale was combined with the subjects’ age, providing the main “non genetic” factor influencing clinical outcome. Subjects were divided into three categories: those having a clinical outcome as expected solely according to age, those having either a worse than expected according to age, and those having a better clinical outcome than expected according to age. The last two were selected as extreme ends of phenotype and used for selecting relevant genes. Second, the method is gene based and processes the genes in a Boolean manner, i. e., having or not having variants. For both common and rare variants, the Boolean classification was repeated three times in order to correspond with subjects having at least the heterozygous genotype (dominant modes), having the homozygous genotype (recessive model), or having the hemizygous genotype (X-linked model) for genes on chromosome X in males and heterozygous genotype for gene on chromosome X ( X-linked dominant model) in females.
Not rarely, genes have more than one coding polymorphism (3413 in the cohort). Various coding changes may move the functioning of a protein either in the same direction (for example lowering or increasing the function) or in the opposite direction, potentially cancelling the effect of juxtaposed genes. We used a “coding haplotype” approach for classifying polymorphic variability in a Boolean manner: we counted the most common combinations (not all theoretical combinations were present due to linkage disequilibrium) and assigned a value of 1 to each gene if the specific combination is present or 0, if the combination was absent. Again, the Boolean classification was repeated three times, corresponding to having at least the heterozygous genotype, having homozygous genotype, or having hemizygous (in males) or heterozygous (in females) genotype for genes on chromosome X. The method treats males and females separately. This represents a first approach to translating into practice sex medicine. As previous studies have borne out, male patients affected by COVID-19 undergo a more severe clinical course. Finally the method we used for gene selection, based on Lasso regularization for a logistic regression classifier, is indirectly confirmed by the association rules produced using a different method.
The proposed method accounts for the role of both common and rare variability, individually and combined. To our knowledge, this is the first method that has been able to synthesize a holistic approach across variabilities, which up until now have been treated separately in host genome research. By using the polygenic score derived from the GWAS approach for the common variability and Mendelian model for rare variability we arrived at a combined approach that significantly outstips the possibility of each of the earlier methodologies approached separately. The introduction of the IPGS formula proved determinative for a significant empowerment of host genome analysis. We call this method the “post-Mendelian model”.
This new method now requires further investigation and refinement. For example, the method treats each gene as having the same weight among common as well as among rare variants. Furthermore, a number of genetic variabilities are missed in the model. These include i) non biallelic common polymorphisms, such as polyamino acid repeats, which are known to contribute to COVID-19 [10]; ii) variability due to germline CNV; iii) variability due to somatic mutations, both CNV and SNP [22, 23]; and iv) very rare variants or private variants whose frequency is too low to be detected by LASSO logistic regression. Further studies that take into account the above reported variability and that increase prediction performances are needed to improve the precision of the model.
5. Conclusion
In conclusion, we were able to identify a set of genes in which common polymorphisms confer either severity or mildness in relation to COVID-19 presentation and development. We also identified another set of genes in which rare variants confer either severity or mildness to COVID-19 disease. We have finally defined a new post-Mendelian genetic model, based on a combined analysis of common and rare variants that appears to explain severity in COVID-19 with a high degree of confidence.
The current version of the is robust enough to extract relevant genes from a specific patient and identify the main pathogenic pathways that are accessible to current personalized co-adjuvant treatment flanking the current use of cortisone. Even in its present stage of development, this post-Mendelian model is strongly capable of identifying candidate genes that can explain with confidence the presentation and development of COVID-19 in patients with mild and severe disease. The proposed model should, however, not be employed immediately as a predictive tool, as it requires more testing, preferably in independent cohorts of patients across various population sets. This model allows us the possibility to summarize and critically evaluate the enormous amount of information now available from sequencing experiments. The immediate link demonstrated here between the gene variants extracted by the automatic analyses and their possible biological roles in COVID-19 supports the idea that this approach to the analysis of genetic information can lead to useful conclusions for COVID-19 diseases management. It is also promising for the further development of this post-Mendelian model with reliable predictive capabilities.
By better understanding the role of host genetics in COVID-19 susceptibility and disease severity, we are also in a stronger position to identify public health measures that will may curb the impact of the disease on society as a whole. This should help us to genetically screen already affected patients as well as, eventually, individuals who may potentially be patients in order to predict those who are more or less susceptible to developing COVID-19 post-infection, especially the more severe cases. It should further help us in, not only reassigning therapeutics or developing new interventions (including vaccines), but also in decision-making regarding therapeutics and vaccine allocations. Beyond what this post-Mendelian method can help us understand regarding the role of host genetics in COVID-19 susceptibility and the potential implications for clinical and public health responses, the model also has strong potential for understanding the role of host genetics in other complex disorders. As we move into an era of precision, patient-centric medicine - an era being propelled by the lurking ubiquitous character of COVID-19 - this post-Mendelian method can help us tailor treatments to the specific needs of individual patients.
Acknowledgments
This study is part of the GEN-COVID Multicenter Study, https://sites.google.com/dbm.unisi.it/gen-covid, the Italian multicenter study aimed at identifying the COVID-19 host genetic bases. Specimens were provided by the COVID-19 Biobank of Siena, which is part of the Genetic Biobank of Siena, member of BBMRI-IT, of Telethon Network of Genetic Biobanks (project no. GTB18001), of EuroBioBank and of D-Connect. We thank the CINECA consortium for providing computational resources and the Network for Italian Genomes (NIG) http://www.nig.cineca.it for its support. We thank private donors for the support provided to A.R. (Department of Medical Biotechnologies, University of Siena) for the COVID-19 host genetics research project (D.L n.18 of March 17, 2020). We also thank the COVID-19 Host Genetics Initiative (https://www.covid19hg.org/). This work was funded by MIUR project “Dipartimenti di Eccellenza 2018-2020” to Department of Medical Biotechnologies University of Siena, Italy (Italian D.L. n.18 March 17, 2020). Private donors for COVID-19 research. “Bando Ricerca COVID-19 Toscana” project to Azienda Ospedaliero-Universitaria Senese (CUP I49C20000280002). Charity fund 2020 from Intesa San Paolo dedicated to the project N. B/2020/0119 “Identificazione delle basi genetiche determinanti la variabilità clinica della risposta a COVID-19 nella popolazione italiana”. the Istituto Buddista Italiano Soka Gakkai for funding the project “PAT-COVID: Host genetics and pathogenetic mechanisms of COVID-19” (ID n. 2020-2016_RIC_3); the Italian Ministry of University and Research for funding within the “Bando FISR 2020” in COVID-19.
Conflicts of interest
All the authors declare no competing financial interests.
Ethics and consent to participate
The GEN-COVID study was consistent with Institutional guidelines and it was approved by the University Hospital of Siena Ethical Review Board (Protocol n. 16929, dated March 16, 2020). As part of GEN-COVID Multicenter Study written informed consent was obtained from all individuals who contributed samples and data. Detailed clinical and laboratory characteristics (data), specifically related to COVID-19, were collected for all subjects.
Consent for publication
The patients were informed of this research and agreed to it through the informed consent process.
Availability of data and materials
The data and samples referenced here are housed in the GEN-COVID Patient Registry and the GEN-COVID Biobank and are available for consultation. You may contact the corresponding author, Prof. Alessandra Renieri (e-mail: alessandra.renieri@unisi.it).
The datasets generated and analysed during the current study are available in the COVID-19 dedicated section (http://nigdb.cineca.it) repository, within the Network for Italian Genome (http://www.nig.cineca.it). There are no restrictions on data access. Only registration is needed.
Institutional review board statement
The GEN-COVID study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the University Hospital of Siena Ethical Review Board (protocol code 16929, dated March 16, 2020).
Author contributions
Conceptualization, Antonio Esposito, Pietro Pinoli, Francesca Mari and Alessandra Renieri; Data curation, Giada Beligni and Mirjam Lista; Formal analysis, Nicola Picchiotti, Elisa Benetti, Chiara Fallerini, Sergio Daga, Margherita Baldassarri, Francesca Fava, Kristina Zguro, Stefano Ceri, Antonio Esposito, Pietro Pinoli, Francis P. Crawley, Elisa Frullanti, GEN-COVID Multicenter Study, Marco Gori, Alessandra Renieri and Simone Furini; Funding acquisition, Antonio Esposito, Pietro Pinoli and Francesca Mari; Methodology, Nicola Picchiotti, lisa Benetti, Floriana Valentino, Gabriella Doddato, Annarita Giliberti, Sara Amitrano, Mirella Bruttini and Simone Furini; Project administration, Antonio Esposito, Pietro Pinoli, Francesca Mari and Alessandra Renieri; Supervision, Antonio Esposito, Pietro Pinoli, Francesca Mari, GEN-COVID ulticenter Study and Alessandra Renieri; Validation, Simone Furini; Writing – original draft, Nicola Picchiotti, Elisa Benetti, Chiara Fallerini, Sergio Daga, Margherita Baldassarri, Francesca Fava, Kristina Zguro, Floriana Valentino, Gabriella Doddato, Annarita Giliberti, Rossella Tita, Sara Amitrano, Mirella Bruttini, Laura Di Sarno, Diana Alaverdian, Giada Beligni, Maria Palmieri, Mirjam Lista, Susanna Croci, Ilaria Meloni, Anna Maria Pinto, Chiara Gabbi, Stefano Ceri, Antonio Esposito, Pietro Pinoli, Francis P. Crawley, Elisa Frullanti, Francesca Mari, GEN-COVID Multicenter Study, Marco Gori, Alessandra Renieri and Simone Furini.
References
- Islam MR, Hoque MN, Rahman MS, et al. Genome-wide analysis of SARS-CoV-2 virus strains circulating worldwide implicates heterogeneity. Sci Rep (2020): 14004.
- Severe Covid-19 GWAS Group, Ellinghaus D, Degenhardt F, et al. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N Engl J Med (2020): 1522-1534.
- Pairo-Castineira E, Clohisey S, Klaric L, et al. Genetic mechanisms of critical illness in Covid-19. Nature 591 (2020): 92-98.
- COVID-19 Host Genetics Initiative et al. Mapping the human genetic architecture of COVID-19. Nature (2021).
- Zhang X, Tan Y, Ling Y, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 583 (2020): 437-440.
- van der Made CI, Simons A, Schuurs-Hoeijmakers J, et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. 324 (2020): 663-673.
- Solanich X, Vargas-Parra G, van der Made CI, et al. Genetic screening for TLR7 variants in young and previously healthy men with severe COVID-19: a case series. Front Immunol (2021): 719115.
- Benetti E, Giliberti A, Emiliozzi A, et al. Clinical and molecular characterization of COVID-19 hospitalized patients. PLoS One 15 (2020): e0242534.
- Daga S, Fallerini C, Baldassarri M, et al. Employing a systematic approach to biobanking and analyzing clinical and genetic data for advancing COVID-19 research. Eur J Hum Genet 29 (2021): 745-759.
- Baldassarri M, Picchiotti N, Fava F, et al. Shorter androgen receptor polyQ alleles protect against life-threatening COVID-19 disease in European males. EBioMedicine (2021): 103246.
- Croci S, Venneri MA, Mantovani S, et al. The polymorphism L412F in TLR3 inhibits autophagy and is a marker of severe COVID-19 in males. In press in Autophagy 2021.
- Fallerini C, Daga S, Benetti E, et al. SELP Asp603Asn and severe thrombosis in COVID-19 males: implication for anti P-selectin monoclonal antibodies treatment. Journal of hematology & oncology 14 (2021): 123.
- Fallerini C, Daga S, Mantovani S, et al. Association of Toll-like receptor 7 variants with life-threatening COVID-19 disease in males: findings from a nested case-control study. Elife (2021): e67569.
- Molnar C. Interpretable-machine-learning. A Guide for Making Black Box Models Explainable (2019).
- Ranjith-Kumar CT, Miller W, Sun J, et al. Effects of single nucleotide polymorphisms on Toll-like receptor 3 activity and expression in cultured cells. J Biol Chem 282 (2007):17696-17705.
- Dhangadamajhi G, Rout R. Association of TLR3 functional variant (rs3775291) with COVID-19 susceptibility and death: a population-scale study. Hum Cell (2021): 1-3.
- Caballero I, Boyd J, Almiñana C, et al. Understanding the dynamics of Toll-like Receptor 5 response to flagellin and its regulation by estradiol. Sci Rep (2017): 40981.
- Georgel AF, Cayet D, Pizzorno A, et al. Toll-like receptor 5 agonist flagellin reduces influenza A virus replication independently of type I interferon and interleukin 22 and improves antiviral efficacy of oseltamivir. Antiviral Res 168 (2019): 28-35.
- Kumar A. Network Proteins of Angiotensin-converting Enzyme 2 but Not Angiotensin-converting Enzyme 2 itself are Host Cell Receptors for SARS-Coronavirus-2 Attachment. Biology, Engineering, Medicine and Science Reports (2021): 01–05.
- Kong Y, Han J, Wu X, et al. VEGF-D: a novel biomarker for detection of COVID-19 progression. Crit Care (2020): 373.
- Shovlin CL, Vizcaychipi MP. Vascular inflammation and endothelial injury in SARS-CoV-2 infection: The overlooked regulatory cascades implicated by the ACE2 gene cluster. QJM. (2020): hcaa241.
- Zekavat SM, Lin SH, Bick AG, et al. Hematopoietic mosaic chromosomal alterations and risk for infection among 767,891 individuals without blood cancer. medRxiv 3 (2020): 100817.
- Bolton KL, Koh Y, Foote MB, et al. Clonal hematopoiesis is associated with risk of severe Covid-19. medRxiv (2020).
Figure S1 Common polymorphisms and rare variants selected by LASSO Logistic Regression. Comparison of extreme ends of phenotype: subjects of class 1 (red in Fig.1) versus class 0 (green in Fig. 1) as defined by ordered logistic regression.
Common (>1%) bi-allelic polymorphisms (coding haplotypes) considering heterozygous variants plus homozygous variants versus wt genotype and using both males and females (S1a) or only males (S1b) or only females (S1c); Common (>1%) bi-allelic polymorphisms (coding haplotypes) considering homozygous variants versus heterozygous variants plus wt genotype and using both males and females (S1d) or only males (S1e) or only in females (S1f) Common (>1%) bi-allelic polymorphisms (coding haplotypes) considering hemizygous variants (only genes on chromosome X) versus wt genotype and using males only (S1g) or females only (S1h);
Rare variants (<1%) considering considering heterozygous variants plus homozygous variants versus wt genotype (dominant model) and using both males and females (S1i) or only males (S1l) or only females (S1m); Rare variants (<1%) considering homozygous variants versus heterozygous variants plus wt genotype (recessive model) and using both males and females (S1n) or only females (S1o). For males (S1p) the performances are below the random guess; Rare variants (<1%) considering hemizygous variants (only genes on chromosome X) versus wt genotype (X-linked model) and using males only (S1q) or only females (S1r). Features are gene-based representations of Genotypic Combinations (GC) of common polymorphisms. The histograms (weights) represented by importance of each feature (genes), inlcuding age and sex, for the classification task (Upper Panel). The positive weights reflect a susceptible behaviour of the gene to the target COVID-19 disease, whereas the negative weights a mildness action. Down Panel: Cross-validation accuracy for the grid of LASSO regularization parameters; the error bar is given by the standard deviation of the average ROC-AUC within the 10 folds; the red point corresponds to the parameter chosen for the fitting procedure.
Supplementary Table 1. Common features extracted in males obtained after 82 bootstraps
Supplementary Table 2. Common features extracted in females obtained after 82 bootstraps
Supplementary Table 3. Rare features extracted in males obtained after 82 bootstraps
Supplementary Table 4. Rare features extracted in females obtained after 82 bootstraps
GEN-COVID Multicenter Study (https://sites.google.com/dbm.unisi.it/gen-covid)
Francesca Montagnani3,10, Mario Tumbarello3,10, Ilaria Rancan3,10, Massimiliano Fabbiani10, Elena Bargagli11, Laura Bergantini11, Miriana D’Alessandro11, Paolo Cameli11, David Bennett11, Federico Anedda12, Simona Marcantonio12, Sabino Scolletta12, Federico Franchi12, Maria Antonietta Mazzei13, Susanna Guerrini13, Edoardo Conticini14, Luca Cantarini14, Bruno Frediani14, Danilo Tacconi15, Chiara Spertilli Raffaelli15, Marco Feri16, Alice Donati16, Raffaele Scala17, Luca Guidelli17, Genni Spargi18, Marta Corridi18, Cesira Nencioni19, Leonardo Croci19, Gian Piero Caldarelli20, Davide Romani21, Paolo Piacentini21 , Maria Bandini21, Elena Desanctis21, Silvia Cappelli21, Anna Canaccini22, Agnese Verzuri22, Valentina Anemoli22, Agostino Ognibene23, Alessandro Pancrazzi23, Maria Lorubbio23, Massimo Vaghi24, Antonella D’Arminio Monforte25, Federica Gaia Miraglia25, Mario U. Mondelli26,27, Stefania Mantovani26, Massimo Girardis28, Sophie Venturelli28, Stefano Busani28, Andrea Cossarizza29, Andrea Antinori30, Alessandra Vergori30, Arianna Emiliozzi30, Stefano Rusconi31,32, Matteo Siano32, Arianna Gabrieli32, Agostino Riva31,32, Daniela Francisci33,34, Elisabetta Schiaroli33, Francesco Paciosi33, Andrea Tommasi33, Pier Giorgio Scotton35, Francesca Andretta35, Sandro Panese36, Stefano Baratti36, Renzo Scaggiante37, Francesca Gatti37, Saverio Giuseppe Parisi38, Francesco Castelli39, Eugenia Quiros-Roldan39, Melania Degli Antoni39, Isabella Zanella40,41, Matteo Della Monica42, Carmelo Piscopo42, Mario Capasso43,44,45, Roberta Russo43,44, Immacolata Andolfo43,44, Achille Iolascon43,44, Giuseppe Fiorentino46, Massimo Carella47, Marco Castori47, Filippo Aucella48, Pamela Raggi49, Carmen Marciano49, Rita Perna49, Matteo Bassetti50,51, Antonio Di Biagio50,51, Maurizio Sanguinetti52,53, Luca Masucci52,53, Alessandra Guarnaccia52, Serafina Valente54, Oreste De Vivo54, Maria Antonietta Mencarelli5, Caterina Lo Rizzo5, Francesca Ariani3,4,5, Marco Mandalà55, Alessia Giorli55, Lorenzo Salerni55, Patrizia Zucchi56, Pierpaolo Parravicini56, Elisabetta Menatti57, Tullio Trotta58, Ferdinando Giannattasio58, Gabriella Coiro58, Fabio Lena59, Leonardo Gianluca Lacerenza59, Cristina Mussini60, Enrico Martinelli61, Luisa Tavecchia62, Mary Ann Belli62, Lia Crotti63,64,65,66,67, Gianfranco Parati63,64, Maurizio Sanarico68, Francesco Raimondi69, Filippo Biscarini70, Alessandra Stella70, Tiziana Bachetti71, Maria Teresa La Rovere72, Maurizio Bussotti73, Serena Ludovisi74, Katia Capitani3,75, Simona Dei76, Sabrina Ravaglia77, Rosangela Artuso78, Elena Andreucci78, Giulia Gori78, Angelica Pagliazzi78, Erika Fiorentin78, Antonio Perrella79, Francesco Bianchi79,3, Paola Bergomi80, Emanuele Catena80, Riccardo Colombo80, Sauro Luchi81, Giovanna Morelli81, Paola Petrocelli81, Sarah Iacopini81, Sara Modica81, Silvia Baroni82, Francesco Vladimiro Segala83, Marco Tanfoni1, Marco Falcone84, Giusy Tiseo84, Chiara Barbieri84, Tommaso Matucci84, Davide Grassi85, Claudio Ferri85, Franco Marinangeli86, Francesco Brancati87, Antonella Vincenti88, Valentina Borgo88, Stefania Lombardi88, Mirco Lenzi88, Massimo Antonio Di Pietro89, Francesca Vichi89, Benedetta Romanin89, Letizia Attala89, Cecilia Costa89, Andrea Gabbuti89, Roberto Menè63,64, Patrizia Casprini90, Giuseppe Merla91,92, Gabriella Maria Squeo91, Marcello Maffezzoni93, Raffaele Bruno94,95, Marco Vecchia94, Marta Colaneri94
- Department of Medical Sciences, Infectious and Tropical Diseases Unit, Azienda Ospedaliera Universitaria Senese, Siena, Italy
- Unit of Respiratory Diseases and Lung Transplantation, Department of Internal and Specialist Medicine, University of Siena, Italy
- Dept of Emergency and Urgency, Medicine, Surgery and Neurosciences, Unit of Intensive Care Medicine, Siena University Hospital, Italy
- Department of Medical, Surgical and Neuro Sciences and Radiological Sciences, Unit of Diagnostic Imaging, University of Siena, Italy
- Rheumatology Unit, Department of Medicine, Surgery and Neurosciences, University of Siena, Policlinico Le Scotte, Italy
- Department of Specialized and Internal Medicine, Infectious Diseases Unit, San Donato Hospital Arezzo, Italy
- Dept of Emergency, Anesthesia Unit, San Donato Hospital, Arezzo, Italy
- Department of Specialized and Internal Medicine, Pneumology Unit and UTIP, San Donato Hospital, Arezzo, Italy
- Department of Emergency, Anesthesia Unit, Misericordia Hospital, Grosseto, Italy
- Department of Specialized and Internal Medicine, Infectious Diseases Unit, Misericordia Hospital, Grosseto, Italy
- Clinical Chemical Analysis Laboratory, Misericordia Hospital, Grosseto, Italy
- Department of Preventive Medicine, Azienda USL Toscana Sud Est, Italy
- Territorial Scientific Technician Department, Azienda USL Toscana Sud Est, Italy
- Clinical Chemical Analysis Laboratory, San Donato Hospital, Arezzo, Italy
- Chirurgia Vascolare, Ospedale Maggiore di Crema, Italy
- Department of Health Sciences, Clinic of Infectious Diseases, ASST Santi Paolo e Carlo, University of Milan, Italy
- Division of Infectious Diseases and Immunology, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
- Department of Internal Medicine and Therapeutics, University of Pavia, Italy
- Department of Anesthesia and Intensive Care, University of Modena and Reggio Emilia, Modena, Italy
- Department of Medical and Surgical Sciences for Children and Adults, University of Modena and Reggio Emilia, Modena, Italy
- HIV/AIDS Department, National Institute for Infectious Diseases, IRCCS, Lazzaro Spallanzani, Rome, Italy
- III Infectious Diseases Unit, ASST-FBF-Sacco, Milan, Italy
- Department of Biomedical and Clinical Sciences Luigi Sacco, University of Milan, Milan, Italy
- Infectious Diseases Clinic, Department of Medicine 2, Azienda Ospedaliera di Perugia and University of Perugia, Santa Maria Hospital, Perugia, Italy
- Infectious Diseases Clinic, "Santa Maria" Hospital, University of Perugia, Perugia, Italy
- Department of Infectious Diseases, Treviso Hospital, Local Health Unit 2 Marca Trevigiana, Treviso, Italy
- Clinical Infectious Diseases, Mestre Hospital, Venezia, Italy.
- Infectious Diseases Clinic, ULSS1, Belluno, Italy
- Department of Molecular Medicine, University of Padova, Italy
- Department of Infectious and Tropical Diseases, University of Brescia and ASST Spedali Civili Hospital, Brescia, Italy
- Department of Molecular and Translational Medicine, University of Brescia, Italy;
- Clinical Chemistry Laboratory, Cytogenetics and Molecular Genetics Section, Diagnostic Department, ASST Spedali Civili di Brescia, Italy
- Medical Genetics and Laboratory of Medical Genetics Unit, A.O.R.N. "Antonio Cardarelli", Naples, Italy
- Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Naples, Italy
- CEINGE Biotecnologie Avanzate, Naples, Italy
- IRCCS SDN, Naples, Italy
- Unit of Respiratory Physiopathology, AORN dei Colli, Monaldi Hospital, Naples, Italy
- Division of Medical Genetics, Fondazione IRCCS Casa Sollievo della Sofferenza Hospital, San Giovanni Rotondo, Italy
- Department of Medical Sciences, Fondazione IRCCS Casa Sollievo della Sofferenza Hospital, San Giovanni Rotondo, Italy
- Clinical Trial Office, Fondazione IRCCS Casa Sollievo della Sofferenza Hospital, San Giovanni Rotondo, Italy
- Department of Health Sciences, University of Genova, Genova, Italy
- Infectious Diseases Clinic, Policlinico San Martino Hospital, IRCCS for Cancer Research Genova, Italy
- Microbiology, Fondazione Policlinico Universitario Agostino Gemelli IRCCS, Catholic University of Medicine, Rome, Italy
- Department of Laboratory Sciences and Infectious Diseases, Fondazione Policlinico Universitario A. Gemelli IRCCS, Rome, Italy
- Department of Cardiovascular Diseases, University of Siena, Siena, Italy
- Otolaryngology Unit, University of Siena, Italy
- Department of Internal Medicine, ASST Valtellina e Alto Lario, Sondrio, Italy
- Study Coordinator Oncologia Medica e Ufficio Flussi Sondrio, Italy
- First Aid Department, Luigi Curto Hospital, Polla, Salerno, Italy
- Department of Pharmaceutical Medicine, Misericordia Hospital, Grosseto, Italy
- Infectious Diseases Clinics, University of Modena and Reggio Emilia, Modena, Italy
- Department of Respiratory Diseases, Azienda Ospedaliera di Cremona, Cremona, Italy
- O.C. Medicina, ASST Nord Milano, Ospedale Bassini, Cinisello Balsamo (MI), Italy
- Istituto Auxologico Italiano, IRCCS, Department of Cardiovascular, Neural and Metabolic Sciences, San Luca Hospital, Milan, Italy
- Department of Medicine and Surgery, University of Milano-Bicocca, Milan, Italy
- Istituto Auxologico Italiano, IRCCS, Center for Cardiac Arrhythmias of Genetic Origin, Milan, Italy
- Istituto Auxologico Italiano, IRCCS, Laboratory of Cardiovascular Genetics, Milan, Italy
- Member of the European Reference Network for Rare, Low Prevalence and Complex Diseases of the Heart-ERN GUARD-Heart
- Independent Data Scientist, Milan, Italy
- Scuola Normale Superiore, Pisa, Italy
- CNR-Consiglio Nazionale delle Ricerche, Istituto di Biologia e Biotecnologia Agraria (IBBA), Milano, Italy
- Direzione Scientifica, Istituti Clinici Scientifici Maugeri IRCCS, Pavia, Italy
- Istituti Clinici Scientifici Maugeri IRCCS, Department of Cardiology, Institute of Montescano, Pavia, Italy
- Istituti Clinici Scientifici Maugeri IRCCS, Department of Cardiology, Institute of Milan, Milan, Italy
- Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, Milan, Italy
- Core Research Laboratory, ISPRO, Florence, Italy
- Health Management, Azienda USL Toscana Sudest, Tuscany, Italy
- IRCCS C. Mondino Foundation, Pavia, Italy
- Medical Genetics Unit, Meyer Children's University Hospital, Florence, Italy
- Department of Medicine, Pneumology Unit, Misericordia Hospital, Grosseto, Italy.
- Department of Anesthesia and Intensive Care Unit, ASST Fatebenefratelli Sacco, Luigi Sacco Hospital, Polo Universitario, University of Milan, Milan
- Infectious Disease Unit, Hospital of Lucca, Italy
- Department of Diagnostic and Laboratory Medicine, Institute of Biochemistry and Clinical Biochemistry, Fondazione Policlinico Universitario A. Gemelli IRCCS, Catholic University of the Sacred Heart, Rome, Italy.
- Clinic of Infectious Diseases, Catholic University of the Sacred Heart, Rome, Italy
- Department of Clinical and Experimental Medicine, Infectious Diseases Unit, University of Pisa, Pisa, Italy
- Department of Clinical Medicine, Public Health, Life and Environment Sciences, University of L'Aquila, Italy
- Anesthesiology and Intensive Care, University of L'Aquila, L'Aquila, Italy
- Medical Genetics Unit, Department of Life, Health and Environmental Sciences, University of L'Aquila, L'Aquila, Italy
- Infectious Disease Unit, Hospital of Massa, Italy
- Unit of Infectious Diseases, S.M. Annunziata Hospital, Florence, Italy..
- Laboratory of Clinical Pathology and Immunoallergy, Florence-Prato, Italy
- Laboratory of Regulatory and Functional Genomics, Fondazione IRCCS Casa Sollievo della Sofferenza, San Giovanni Rotondo (Foggia), Italy
- Department of Molecular Medicine and Medical Biotechnology, University of Naples Federico II, Naples, Italy.
- University of Pavia, Pavia, Italy
- Division of Infectious Diseases I, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
- Department of Clinical, Surgical, Diagnostic, and Pediatric Sciences, University of Pavia, Pavia, Italy

 Impact Factor: * 5.6
Impact Factor: * 5.6 Acceptance Rate: 74.36%
Acceptance Rate: 74.36%  Time to first decision: 10.4 days
Time to first decision: 10.4 days  Time from article received to acceptance: 2-3 weeks
Time from article received to acceptance: 2-3 weeks